




CRISPR-Cas9, often shortened to CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats), is a revolutionary gene-editing technology that has transformed the landscape of biological research and therapeutic development.
It functions like a pair of highly precise molecular scissors, capable of finding a specific sequence of DNA within a cell and making a cut or modification at that exact location.
The system is guided by a piece of RNA (guide RNA) that is designed to match the target DNA sequence, leading the Cas9 enzyme (the “scissors”) to the correct spot in the genome (Innovative Genomics Institute, n.d.).
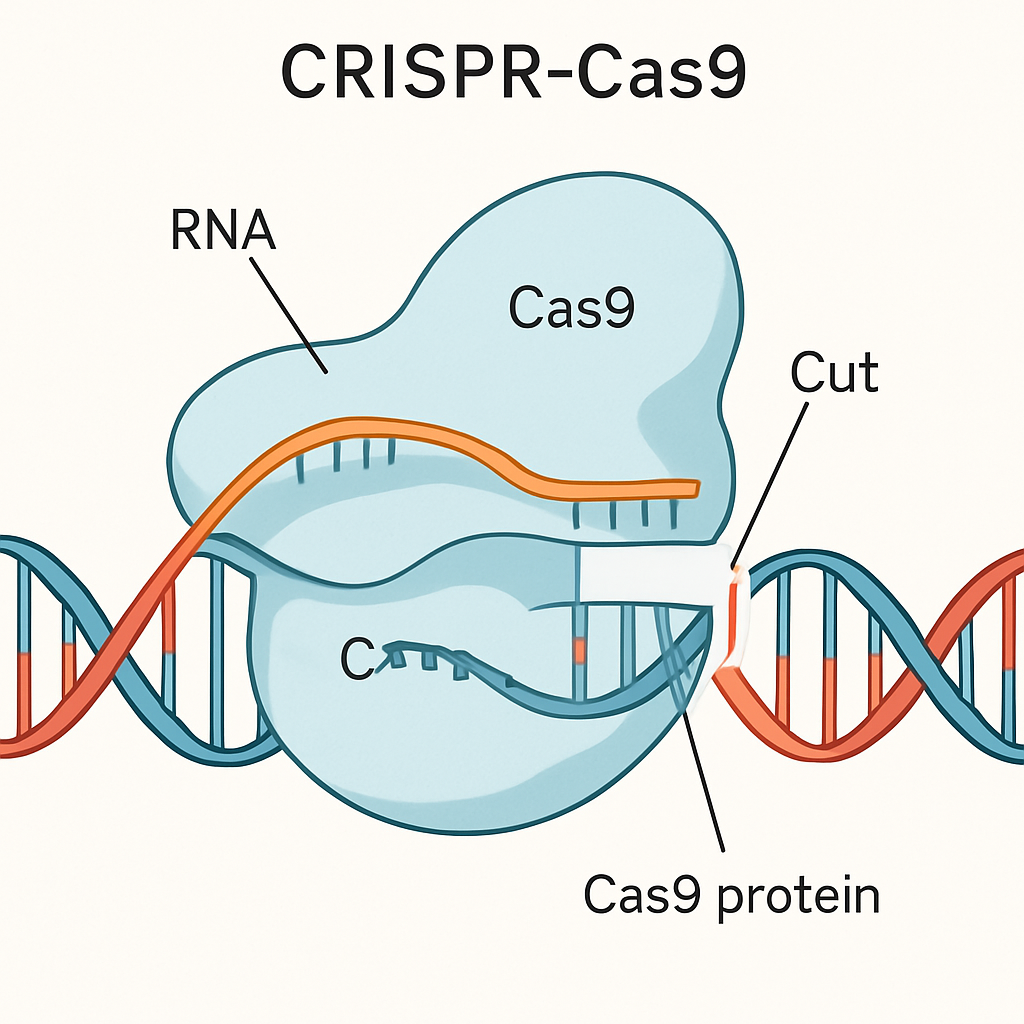 Conceptual illustration of the CRISPR-Cas9 system editing a DNA strand. This technology allows for precise modifications to the genetic code.
Conceptual illustration of the CRISPR-Cas9 system editing a DNA strand. This technology allows for precise modifications to the genetic code.In the case of KJ Muldoon, researchers at CHOP and Penn Medicine embarked on an unprecedented journey: to develop a personalized CRISPR-based therapy. This wasn’t a pre-existing, off-the-shelf-treatment. Instead, it was custom-designed to correct the specific mutation in KJ’s CPS1 gene. This approach, sometimes referred to as “n-of-1” therapy (a treatment developed for a single individual), represents a significant departure from traditional drug development, which typically targets larger patient populations (Children’s Hospital of Philadelphia, 2025).
The team, led by Dr. Rebecca Ahrens-Nicklas of CHOP and Dr. Kiran Musunuru of Penn Medicine, leveraged their extensive expertise in rare metabolic disorders and gene editing. They focused on a type of CRISPR technology called base editing. Unlike standard CRISPR-Cas9 which cuts both strands of the DNA, base editing makes a precise chemical change to a single DNA letter (base) without causing a double-strand break. This can be a safer approach as it reduces the risk of unintended genetic changes (off-target effects) or larger DNA rearrangements that can sometimes occur with double-strand breaks.
Remarkably, the entire process, from identifying KJ’s specific mutation to designing, manufacturing, and administering the first dose of the bespoke base editing therapy, was accomplished in just six to seven months (Children’s Hospital of Philadelphia, 2025; CBS News, 2025). This rapid development is a testament to the advancements in gene-editing technology and the collaborative spirit of the researchers and clinicians involved. The therapy was delivered to KJ’s liver – the primary site of the CPS1 enzyme’s function – using lipid nanoparticles (LNPs). LNPs are tiny fatty spheres that can encapsulate the gene-editing machinery and ferry it into the target liver cells. Once inside the cells, the base editor was designed to find KJ’s mutated CPS1 gene and correct the single incorrect DNA letter, thereby restoring the gene’s ability to produce a functional CPS1 enzyme.
KJ received his first infusion of the experimental therapy in February 2025, followed by subsequent doses. The careful planning and execution of this novel treatment underscore the meticulous approach required when venturing into such uncharted medical territory.

